This guide provides step-by-step instructions for depositing a simple thin-film morphology and performing electronic structure analysis on it using Nanoscope. It is designed to help you quickly get started with the basic functionalities of Nanoscope. For more complex use cases, please refer to the User Guide section.
Below, we provide settings for each module in the Nanoscope workflow.
Each module includes configurations for two types of runs: Production Runs for meaningful and accurate results,
and Test Runs for quick checks of workflow functionality and output.
Production Runs
Test Runs
Settings for production runs, resulting in meaningful, accurate results.
Settings suitable for quick technical tests that deliver quick (but meaningless) results also on small computational resources.
Refer to the installation guide for recommendations for hardware and software setup for production runs and quick technical tests.
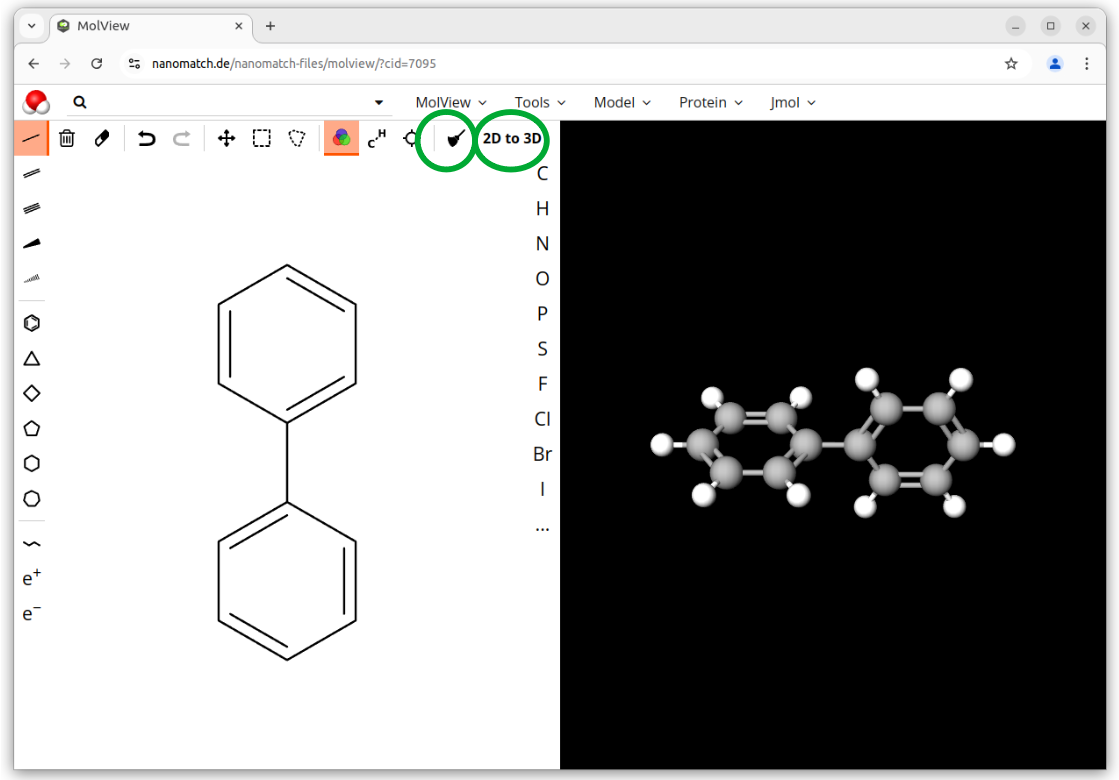
We use biphenyl as a simple example as it allows for quick computation. It is not meant as a physical case study. Feel free to try a different molecule. Keep in mind that the basic usage of Nanoscope covers molecules with up to 40 atoms.
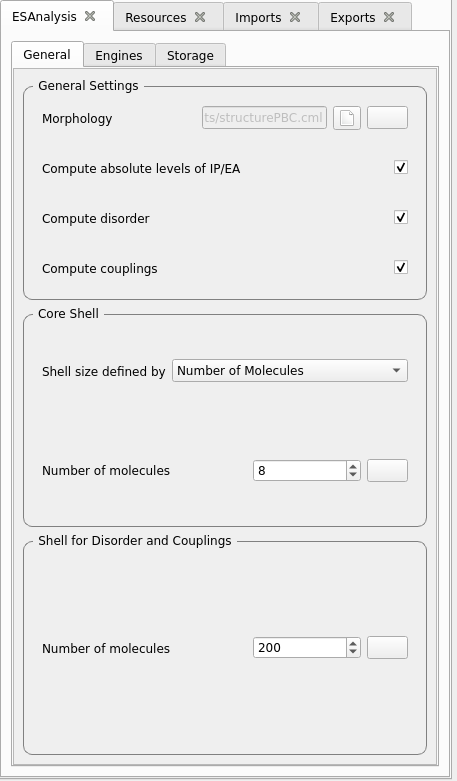
Drag&Drop the modules MolPrep, Deposit and ESAnalysis from the top left panel into the middle workflow panel into a linear workflow and arrange as depicted below. Double click on each module to adapt settings and allocate resources for each simulation step.
In the central panel, double-click on the module to set it up.
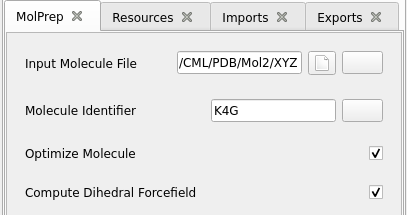
MolPrep.
Set the Input Molecule File: select the molecule you created above.
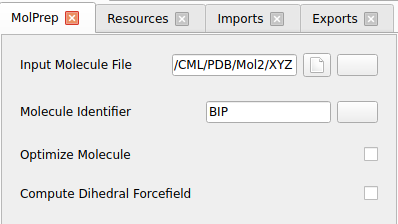
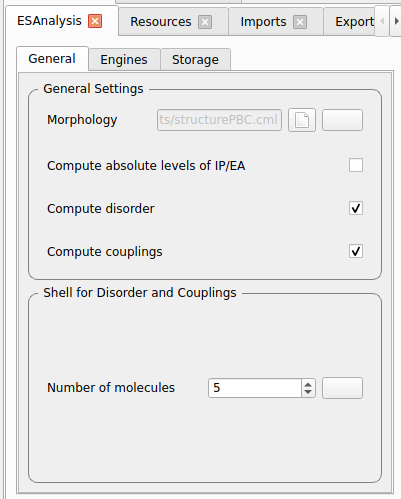
Only for test runs:
Disable Optimize Molecule
Disable Compute Dihedral Forcefield
Production runs
Test runs
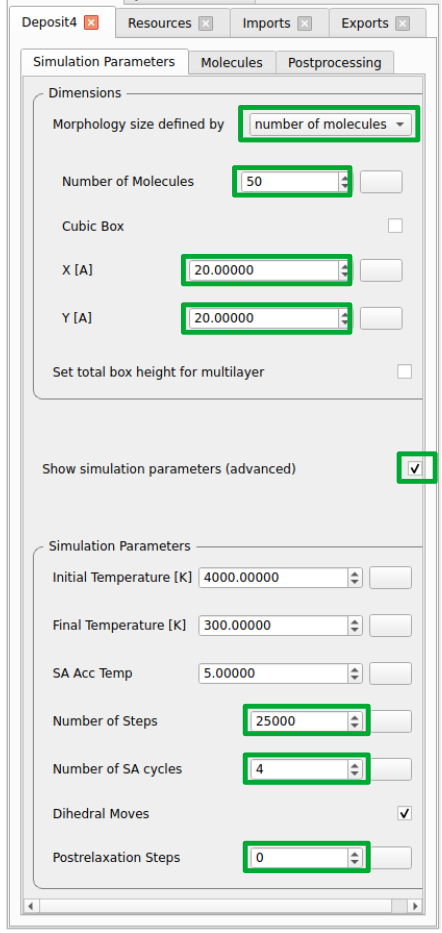
Deposit
Adjust the SimulationParameters tab as indicated in the screenshots below. Note that for a standard production run, the preset simulation parameters can be used without adjustment.
Production runs
Test runs
In the Molecules tab:
Click on the rightmost buttons next to the input fields to load molecule and forcefield file from MolPrep:
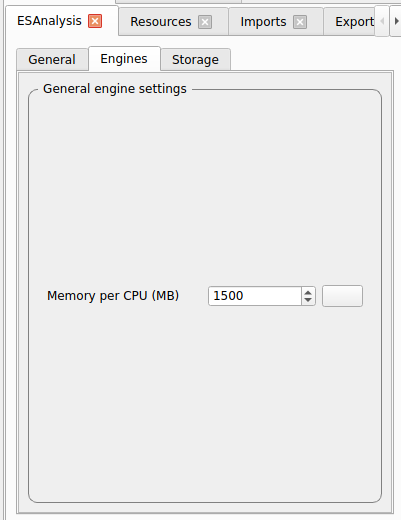
For each module, go to the Resources tab and set the computational resources:
For test runs using test-settings as indicated above: Use whatever you have available
For production runs, the following is recommended:
Module
CPUs
Memory (MB)
Walltime
MolPrep
≥32
≥64000
A few
hours
Deposit
32
≥64000
A few
hours
ESAnalysis
≥64
≥128000
Several
hours
Please do not forget to set up resources for each and every module in your workflow as animated below.
Note
You can run the workflow with fewer cores, if the above resources are not available. This increases runtime respectively.
Memory is provided in MB in the Resources tab. Running Nanoscope with less memory than indicated in the table above is possible, but you may run into out-of-memory issues especially for larger molecules.
Walltime is provided in seconds in the Resources tab.
The primary outputs of the ESAnalysis module are located in the Analysis/DOS directory within the module’s runtime folder.
HOMO and LUMO distribution in a pristine morphology. The values in the figure are onsets of the distributions that compare to experimental values.
Further outputs are:
File
Content
DOS_Gaussian.png
Plot visualizing the Gaussian-broadened density of HOMO and LUMO levels without vibrational effects.
Vibrational_Gaussian_DOS_plot.png
Plot showing the Gaussian-broadened HOMO/LUMO distribution including vibrational broadening.
all_DOS_plot.png
Combined plot overlaying DOS distributions with and without vibrational broadening (both are Gaussian-broadened).
raw_data_homo_lumo.yaml
Exact HOMO and LUMO energies (in mixed morphologies for each molecule type). Includes mean, std, and all individual energy levels.
homo_lumo_onsets.yaml
Calculated onset energies for HOMO and LUMO levels distribution for each molecule type, can be compared with experimental onsets.
homo_lumo_centers.yaml
Mean and standard deviation of the distribution of HOMO and LUMO levels for each molecule type. Can be used as an ab-initio input for multi-scale simulation workflows.